Sull’uso off-label dell’idrossiclorochina per il trattamento del COVID-19 (nota a Consiglio di Stato, Sezione Terza, ord.za 11 dicembre 2020, n. 7097) di Giordana Strazza
Sommario: 1. Premessa - 2. L’uso off-label dei farmaci- 3. Le novità sulla sperimentazione dei medicinali introdotte per l’emergenza COVID-19 - 4. L’uso off-label dell’idrossiclorochina per il trattamento del COVID-19; 5. L’ordinanza cautelare del Consiglio di Stato; 6. Osservazioni a prima lettura - 7. Conclusioni.
1. Premessa
La pandemia, dovuta alla progressiva diffusione del COVID-19[1], ha posto la comunità scientifica di fronte all’esigenza impellente di individuare terapie farmacologiche sperimentali, per tentare di arginare le conseguenze (non solo sanitarie) causate dal nuovo virus.
Nelle more del vaccino contro il COVID-19[2], infatti, la sperimentazione di farmaci[3] già in commercio per il trattamento di altre malattie ha ricoperto (almeno in potenza) un ruolo-chiave per limitare gli effetti del virus.
Le incertezze scientifiche sull’utilizzo sperimentale di terapie farmacologiche – in particolare, dell’idrossiclorochina[4]– per il trattamento del COVID-19 hanno avuto, però, rilevanti ripercussioni anche sul piano giuridico.
La parabola sull’uso dell’idrossiclorochina per i pazienti con COVID-19 ha innescato, infatti, un “braccio di ferro” tra l’Agenzia italiana del farmaco (AIFA)[5] e il Consiglio di Stato, al punto tale da riportare in auge il tema “classico” del sindacato sulla discrezionalità tecnica.
L’AIFA – che fa parte del sistema normativo europeo per i medicinali, costruito sul modello a rete, che coinvolge le autorità di regolazione nazionali dello Spazio economico europeo, l’Agenzia europea per i medicinali (EMA) e la Commissione europea – svolge, infatti, un ruolo centrale in termini di farmacovigilanza, di sperimentazione e di ricerca medicinale, oltre che di informazione pubblica[6], sulla base dei dati forniti dalla scienza di settore.
2. L’uso off-label dei farmaci
Come evidenziato, per contrastare la pandemia dovuta al COVID-19, sono state riposte grandi speranze sull’uso off-label di alcuni medicinali, ossia sull’impiego nella pratica clinica di farmaci già registrati, ma utilizzati in maniera non conforme (per patologia, popolazione o posologia)[7] da quanto previsto dall’autorizzazione all’immissione in commercio (AIC)[8].
Il ricorso ai farmaci off-label riguarda, dunque, molecole ampiamente note che, però, sulla base di nuove evidenze scientifiche, possono essere usate, in modo razionale, anche in situazioni cliniche non previste nella scheda tecnica e nel foglietto illustrativo.
L’esigenza è, quindi, quella di bilanciare le opportunità di cura e i progressi nella conoscenza e nella terapia di alcune patologie offerti dalle prescrizioni “fuori indicazione” (artt. 9 e 33 Cost.) e la necessità di preservare i pazienti da rischi per la loro salute (art. 32 Cost.).
In Italia, l’impiego di farmaci “fuori dal bugiardino” [9] è sottoposto a una disciplina piuttosto articolata e disorganica.
Nel nostro ordinamento, nel tentativo di realizzare il bilanciamento di interessi sopra accennato, il d.l. 21 ottobre 1996, n. 536 (recante “Misure per il contenimento della spesa farmaceutica e la rideterminazione del tetto di spesa per l’anno 1996”), convertito nella legge 23 dicembre 1996, n. 648, ha ammesso, a certe condizioni, la possibilità di prescrivere e di utilizzare farmaci al di fuori delle indicazione terapeutiche approvate[10].
L’art. 1, comma 4, della legge appena citata ha stabilito, infatti, che – in assenza di valida alternativa terapeutica[11] – sono erogabili a totale carico del Sistema sanitario nazionale (SSN), i medicinali innovativi la cui commercializzazione è autorizzata in altri Stati, ma non sul territorio nazionale (un-licensed use); quelli non ancora autorizzati, ma sottoposti a sperimentazione clinica (compassionate use); quelli da impiegare per un’indicazione terapeutica diversa da quella autorizzata (off-label use), purché inseriti in un apposito elenco predisposto e periodicamente aggiornato dalla Commissione tecnico-scientifica dell’AIFA, in conformità alle procedure e ai criteri a suo tempo adottati dalla Commissione unica del farmaco (CUF) [12].
Secondo tale disciplina, dunque, il medico che prescrive un farmaco inserito nella c.d. lista 648 deve rispettare le condizioni ivi indicate, con dichiarazione di assunzione di responsabilità del trattamento, previa acquisizione del consenso informato dal paziente.
L’excursus normativo sulle prescrizioni off label include anche l’art. 3 del decreto-legge 17 febbraio 1998, n. 23, convertito dalla legge 8 aprile 1998, n. 94 (“legge Di Bella”)[13] ai sensi del quale, di regola, nel prescrivere un farmaco, il medico deve attenersi “alle indicazioni terapeutiche, alle vie e alle modalità di somministrazione previste dall'autorizzazione all'immissione in commercio” riferibile allo stesso medicinale.
L’art. 3, comma 2, della stessa legge ha individuato, però, un’eccezione a tale regola generale, circoscritta nei seguenti termini: “in singoli casi il medico può, sotto la sua diretta responsabilità e previa informazione del paziente e acquisizione del consenso dello stesso, impiegare il medicinale prodotto industrialmente per un’indicazione o una via di somministrazione o una modalità di somministrazione o di utilizzazione diversa da quella autorizzata, ovvero riconosciuta agli effetti dell’applicazione dell’art. 1, comma 4, del D.L. 21 ottobre 1996, n. 536, convertito dalla Legge 23 dicembre 1996, n. 648, qualora il medico stesso ritenga, in base a dati documentabili, che il paziente non possa essere utilmente trattato con medicinali per i quali sia già approvata quell’indicazione terapeutica o quella via o modalità di somministrazione e purché tale impiego sia noto e conforme a lavori apparsi su pubblicazioni scientifiche accreditate in campo internazionale”[14].
Ad ogni modo, l’art. 3 comma 4, della legge appena citata ha precisato che tale possibilità non costituisce riconoscimento del diritto del paziente all’erogazione dei medicinali a carico del SSN, al di fuori dell’ipotesi disciplinata dall’art. 1, comma 4, della l. n. 648/1996[15].
A ciò si aggiunga che il d.m. 8 maggio 2003, sull’ “Uso terapeutico di medicinali in sperimentazione clinica”, ha regolamentato il c.d. “uso compassionevole” dei farmaci, per consentire (entro determinati limiti) l’accesso a terapie farmacologiche sperimentali ai pazienti privi di alternative terapeutiche, con oneri a carico delle imprese produttrici.
Il decreto sopra indicato ha previsto, infatti, che un medicinale sottoposto a sperimentazione clinica in Italia o all’estero possa essere richiesto all’impresa produttrice per l’uso al di fuori della sperimentazione clinica “qualora non esista valida alternativa terapeutica al trattamento di patologie gravi o di malattie rare o di condizioni di malattia che pongono il paziente in pericolo di vita”.
Secondo tale d.m, l’autorizzazione all’uso è rilasciata solo alle condizioni seguenti: il medicinale sia oggetto, per la medesima indicazione terapeutica, di studi clinici, in corso o conclusi, di fase terza[16] o, in caso di condizioni di malattia che pongono il paziente in pericolo di vita, di fase seconda, già conclusi; i dati disponibili sulle sperimentazioni siano sufficienti per formulare un favorevole giudizio sull’efficacia e sulla tollerabilità del medicinale (ferma l’approvazione del protocollo terapeutico da parte del Comitato Etico nel cui ambito ha avuto origine la richiesta e la notifica al Ministero della salute, i cui uffici possono formulare un eventuale giudizio sospensivo della procedura o dell’uso).
Nella realtà ospedaliera, però, l’utilizzo dei farmaci fuori indicazione spesso non era relegato al rango dell’eccezionalità, ma assumeva la portata di una vera e propria “prassi quotidiana”[17].
Per ovviare a tale impiego improprio dell’off-label è intervenuto l’art. 1, comma 796, lett. z), delle legge 27 dicembre 2006, n. 296 (c.d. legge Finanziaria del 2007).
Tale norma ha disposto, infatti, che la fattispecie prevista dall’art. 3, comma 2, della c.d. legge Di Bella “non sia applicabile al ricorso a terapie farmacologiche a carico del SSN, che, nell’ambito dei presidi ospedalieri o di altre strutture e interventi sanitari, assuma carattere diffuso e sistematico e si configuri, al di fuori delle condizioni di autorizzazione all’immissione in commercio, quale alternativa terapeutica rivolta a pazienti portatori di patologie per le quali risultino autorizzati farmaci recanti specifica indicazione al trattamento”.
La ratio di tale disposizione consiste, infatti, nel prevenire l’abuso di farmaci impiegati off-label, così da evitare l’utilizzo indiscriminato di medicinali senza adeguata verifica di indicazioni terapeutiche da parte delle Agenzie regolatorie.
A distanza di un anno, l’art. 2, comma 348, della legge 24 dicembre 2007, n. 244 (c.d. legge Finanziaria 2008) ha previsto, inoltre, che ogni somministrazione off-label da parte del medico curante richieda “almeno dati favorevoli di sperimentazione clinica di fase seconda”, “introducendo così un elemento di oggettività riconoscibile secondo criteri scientifici certi e riconosciuti”[18].
Il successivo comma 349 ha soggiunto anche che, ai fini delle decisioni sui farmaci off-label, la Commissione tecnico-scientifica dell'AIFA valuta, “oltre i profili di sicurezza, la presumibile efficacia del medicinale, sulla base dei dati disponibili delle sperimentazioni cliniche già concluse, almeno di fase seconda”.
Il 2 dicembre 2017 è poi entrato in vigore il nuovo decreto ministeriale sulla “Disciplina l'uso terapeutico di un medicinale sottoposto a sperimentazione clinica”, che ha abrogato il citato d.m. 8 maggio 2003, sugli usi compassionevoli, e ha previsto nuove norme al riguardo, in conformità dell’art. 158, co. 10, d.lgs. 24 aprile 2006, n. 219[19](“Attuazione della direttiva 2001/83/CE (e successive direttive di modifica) relativa ad un codice comunitario concernente i medicinali per uso umano, nonchè della direttiva 2003/94/CE”).
3. Le novità sulla sperimentazione dei medicinali introdotte per l’emergenza COVID-19
Per l’esigenza di fronteggiare l’emergenza sanitaria dovuta al SARS-CoV-2, l’art. 17 (“Disposizioni urgenti materia di sperimentazione dei medicinali e dispositivi medici per l’emergenza epidemiologica da Covid-19”) del d.l. 17 marzo 2020, n. 18 (“Misure di potenziamento del Servizio sanitario nazionale e di sostegno economico per famiglie, lavoratori e imprese connesse all’emergenza epidemiologica da Covid-19”), come convertito nella legge 24 aprile 2020, n. 27, ha introdotto delle novità sulla sperimentazione dei farmaci per pazienti che hanno contratto il nuovo virus[20].
Tale norma ha stabilito, infatti, che “Limitatamente al periodo dello stato di emergenza, […] ferme restando le disposizioni vigenti in materia di sperimentazione clinica dei medicinali e dei dispositivi medici, al fine di migliorare la capacità di coordinamento e di analisi delle evidenze scientifiche disponibili, è affidata ad Aifa, la possibilità di accedere a tutti i dati degli studi sperimentali e degli usi compassionevoli”.
Al contempo, la disciplina emergenziale, ha istituto un Comitato etico unico centrale, a livello nazionale, in capo a quello già afferente Istituto nazionale per le malattie infettive Lazzaro Spallanzani (IRCCS), per valutare le sperimentazioni farmaceutiche intraprese nelle aziende ospedaliere italiane e instaurare un dialogo con l’Agenzia italiana del farmaco.
Nell’ambito dell’emergenza epidemiologica, dunque, sono state apportate delle deroghe (temporanee) a quanto previsto dal d.lgs. 6 novembre 2007, n. 200 (recante “Principi e linee guida dettagliate per la buona pratica clinica relativa ai medicinali in fase di sperimentazione a uso umano, nonché requisiti per l’autorizzazione alla fabbricazione o importazione di tali medicinali”)[21], finalizzate a una semplificazione e centralizzazione della procedura, e all’AIFA è stato affidato un ruolo-chiave nel controllo (preventivo)[22] sulle sperimentazioni cliniche sui medicinali per pazienti con COVID-19[23].
L’Autorità ha tentato, quindi, di far fronte all’emergenza sanitaria con procedure straordinarie e semplificate per la presentazione e l’approvazione delle sperimentazioni e per la definizione delle modalità di adesione agli studi e di acquisizione dei dati[24].
Al contempo, l’Agenzia italiana del farmaco ha costituito un’apposita unità di crisi, impegnata – tra l’altro – sui farmaci in uso "off label"[25].
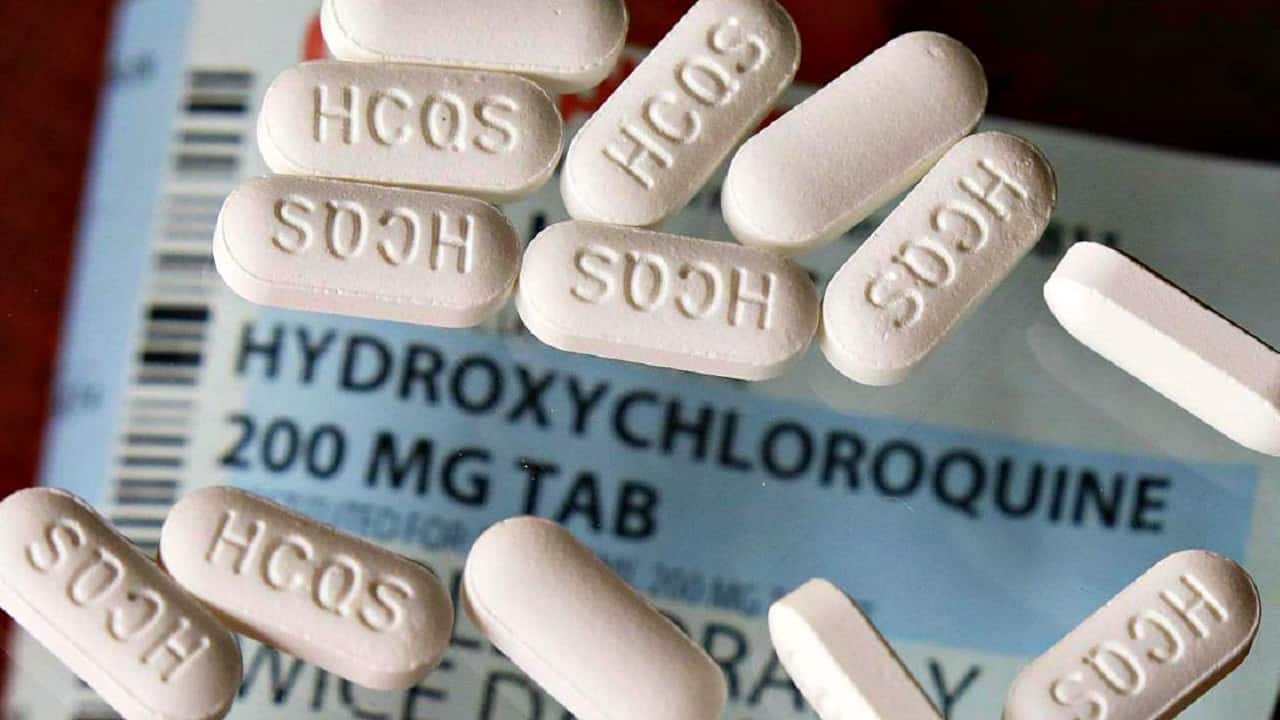
4. L’uso off-label dell’idrossiclorochina per il trattamento del COVID-19
In un primo momento, con nota del 17 marzo 2020, l’AIFA ha comunicato che la sua Commissione Tecnico Scientifica aveva espresso parere favorevole in merito, tra l’altro, all’inserimento a carico del SSN (in deroga alla l. n. 648/1996[26]) dell’uso off-label dell’idrossiclorochina per il trattamento dell’infezione da SARS-CoV-2[27].
Due settimane dopo, l’Agenzia ha pubblicato una nota informativa, in cui sono indicate le informazioni di sicurezza e le principali interazione farmacologiche dell’idrossiclorochina[28].
In quell’occasione, l’AIFA ha evidenziato la comparsa di casi di cardiotossicità e ha raccomandato, prima della prescrizione, di prestare attenzione, con particolare riferimento ai soggetti con disturbi della conduzione cardiaca, con favismo o in presenza di altre terapie concomitanti.
Nello stesso giorno, l’Agenzia europea per i medicinali ha comunicato che “al momento e sulla base dei dati preliminari presentati” “nessun farmaco ha ancora dimostrato la sua efficacia nel trattamento del COVID-19” e ha soggiunto che tra i potenziali trattamenti per il COVID-19, sottoposti a sperimentazione clinica per valutare la sicurezza ed efficacia contro la malattia, era incluso l’utilizzo dell’idorossiclorochina[29].
Pochi giorni dopo, l’Agenzia europea ha sottolineato che la clorochina e l’idrossiclorochina devono essere utilizzati solo negli studi clinici o nei programmi di utilizzo in emergenza per il trattamento del COVID-19[30]. Si legge, infatti, che i due medicinali “sono oggetto di studio in tutto il mondo in quanto potenzialmente in grado di curare la malattia da coronavirus (COVID-19). Tuttavia, l'efficacia nel trattamento del COVID19 non è ancora stata dimostrata negli studi”. Per di più, entrambi i medicinali “possono avere effetti indesiderati gravi, soprattutto a dosi elevate o in associazione ad altri farmaci.
Nel frattempo, l’Agenzia italiana del farmaco ha autorizzato una serie di sperimentazione cliniche per valutare l’efficacia dell’idrossiclorochina, rispetto allo standard di cura, per il trattamento domiciliare di pazienti con un quadro clinico lieve di COVID-19 e in isolamento domiciliare[31].
Il 23 aprile, l’Agenzia europea per i medicinali ha richiamato nuovamente l’attenzione sul rischio di gravi effetti indesiderati con clorochina e idrossiclorochina impiegati nel contesto della pandemia in corso per il trattamento di pazienti con COVID-19[32].
Qualche giorno dopo, ossia il 29 aprile 2020, l’AIFA ha aggiornato la scheda sull’utilizzo di idrossiclorochina per la terapia dei pazienti affetti da COVID-19[33], con una revisione critica delle ultime evidenze di letteratura e con le prove di efficacia e sicurezza disponibili al momento, fornendo ai clinici elementi utili a orientare la prescrizione e a definire un rapporto fra i benefici e i rischi sul singolo paziente.
Il 26 maggio 2020, dato che “nuove evidenze cliniche relative all’utilizzo di idrossiclorochina nei soggetti con infezione da SARS-CoV-2 (seppur derivanti da studi osservazionali o da trial clinici di qualità metodologica non elevata) indicano un aumento di rischio per reazioni avverse a fronte di benefici scarsi o assenti … in attesa di ottenere prove più solide dagli studi clinici in corso in Italia e in altri Paesi (con particolare riferimento a quelli randomizzati)”, l’AIFA ha sospeso l’autorizzazione all’utilizzo di idrossiclorochina per il trattamento dell’infezione da SARS-CoV-2, “al di fuori degli studi clinici, sia in ambito ospedaliero che in ambito domiciliare. Tale utilizzo viene conseguentemente escluso dalla rimborsabilità”[34].
Il 29 maggio, con apposita scheda, l’AIFA ha indicato le motivazioni alla base della decisione di sospendere l’autorizzazione all’utilizzo di idrossiclorochina (e di clorochina) per il trattamento del COVID-19 al di fuori degli studi clinici[35].
Lo stesso giorno l’EMA ha ribadito i rischi (principalmente cardiaci e neuropsichiatrici) connessi all’uso del farmaco nei pazienti COVID-19, per come emersi da studi osservazionali, e ha evidenziato che, “alla luce dei dati emergenti, alcuni paesi dell'UE hanno sospeso o interrotto le sperimentazioni cliniche”[36].
Nel comunicato, si legge, inoltre, che “Per alcune sperimentazioni, tra cui il grande studio multinazionale Solidaritydell’OMS, è stato sospeso l’arruolamento dei pazienti nei bracci che prevedono l’impiego di questi medicinali[37]. La revisione preliminare di Recovery, uno studio in corso di grandi dimensioni sui pazienti con COVID-19, non ha individuato motivi per sospendere o interrompere la sperimentazione”[38].
Le sospensioni sono state adottate, peraltro, dopo la pubblicazione di uno studio sulla prestigiosa rivista “The Lancet”, poi ritirato, che ha evidenziato l’(in)efficacia e i rischi del farmaco per il trattamento del COVID-19[39].
“Alla luce delle attuali evidenze di letteratura”, il 22 luglio 2020, l’AIFA ha confermato “la sospensione dell’autorizzazione all’utilizzo off-label dell’idrossiclorochina al di fuori degli studi clinici”[40].
Ne è sorta una querelle nella stessa comunità scientifica-medica, prima ancora che in ambito giuridico.
A coloro che hanno affermato l’inefficacia dell’idrossiclorochina per i pazienti con COVID-19[41] si sono contrapposti i sostenitori dell’impiego del farmaco (tendenzialmente nella fase precoce di sintomatologia)[42] al punto che la rivista Panorama ha lanciato una petizione (che ha ormai superato le quindicimila firme) per chiedere all'Agenzia italiana del farmaco di “ripristinare l'uso dell'idrossiclorochina”.
5. L’ordinanza cautelare del Consiglio di Stato
A seguito del ricorso proposto da un gruppo di medici avverso la citata sospensione dell’AIFA, la “parabola” sull’uso dell’idrossiclorochina per il trattamento dei pazienti con COVID-19 è proseguita dinanzi al giudice amministrativo.
Con l’(approfondita) ordinanza 11 dicembre 2020, n. 7097, la terza Sezione del Consiglio di Stato ha accolto l’appello cautelare proposto dai ricorrenti[43] e ha sospeso la nota (del 22 luglio) dell’Autorità italiana del farmaco[44].
Secondo il Collegio, innanzitutto, a differenza di quanto sostenuto nella memoria dell’AIFA, l’atto impugnato non si limita a sospendere l’autorizzazione all’utilizzo off-label del farmaco al di fuori degli studi clinici randomizzati controllati (e ad escluderne la rimborsabilità a carico del SSN), ma limita la libertà prescrittiva del medico, vietando l’utilizzo dell’idrossiclorochina[45].
Il Consiglio di Stato ha soggiunto che la c.d. riserva di scienza dell’AIFA non la sottrae al sindacato giurisdizionale del giudice amministrativo sul corretto esercizio della discrezionalità tecnica, basato sulla conoscenza piena del fatto e del percorso intellettivo e volitivo seguito dall’Amministrazione.
Pur riconoscendo il “delicato compito” affidato alla citata Autorità e pur non potendo “decretare l’efficacia terapeutica dell’idrossiclorochina nel contrasto al SARS-CoV-2 in una fase iniziale della malattia, proprio per i limiti connaturati al suo sindacato giurisdizionale”, il Consiglio di Stato ha affermato “il dovere di rilevare che la perdurante incertezza circa la sua efficacia terapeutica, ammessa dalla stessa AIFA a giustificazione dell’ulteriore valutazione in studi clinici randomizzati, non è ragione sufficiente sul piano giuridico a giustificare l’irragionevole sospensione del suo utilizzo sul territorio nazionale da parte dei medici curanti in base ad una conclusione – la totale definitiva inefficacia del farmaco sotto ogni aspetto, anche immunomodulatorio – che, allo stato delle conoscenze e della ricerche tuttora parziali e provvisorie, sembra radicale e prematura già a livello scientifico”.
Secondo il Collegio, dunque, “La scelta se utilizzare o meno il farmaco, in una situazione di dubbio e di contrasto nella comunità scientifica, sulla base di dati clinici non univoci, circa la sua efficacia nel solo stadio iniziale della malattia, deve essere dunque rimessa all’autonomia decisionale e alla responsabilità del singolo medico, con l’ovvio consenso informato del singolo paziente, e non ad una astratta affermazione di principio, in nome di un modello scientifico puro, declinato da AIFA con un aprioristico e generalizzato, ancorché temporaneo, divieto di utilizzo”.
6. Osservazioni a prima lettura
La premessa di fondo su cui si snoda l’ordinanza cautelare, secondo cui la c.d. riserva di scienza non sottrae l’atto di un’autorità amministrativa al sindacato del giudice amministrativo, è – in linea teorica – assodata e inconfutabile, in virtù di quanto sancito dagli artt. 24, 111 e 113 Cost., oltre che dall’art. 47 della Carta dei diritti fondamentali dell’U.e. e dagli artt. 6, par. 1, 7 e 13 della CEDU[46].
In concreto, però, è estremamente sottile e labile la linea di confine del (consentito – rectius dovuto – e costituzionalmente sancito) controllo giurisdizionale di legittimità degli atti che, secondo quanto evidenziato dallo stesso Collegio, sono esercizio di discrezionalità tecnica[47].
Come noto, quest’ultima espressione – considerata “elegante ossimoro”[48] – concerne le valutazioni compiute dalla pubblica Amministrazione quando l’esame dei fatti implica il ricorso a cognizioni tecniche e scientifiche di carattere specialistico, che non appartengono al diritto e che non conducono a un risultato univoco.
I giudizi formulati sulla base di regole tecniche/scientifiche inesatte sono connotati, infatti, dall’opinabilità, tenuta tradizionalmente distinta dall’opportunità[49]: la discrezionalità tecnica, a differenza di quella amministrativa o “pura”, non dovrebbe implicare una scelta (di merito), ma per l’appunto, un giudizio[50].
Posta tale distinzione, grazie all’opera della giurisprudenza amministrativa[51], e alle novità introdotte per effetto della l. 2000, n. 205, che ha previsto, tra l’altro, la consulenza tecnica nel processo amministrativo[52], il sindacato giurisdizionale sulle valutazioni tecniche non si è arrestato al mero controllo formale ed estrinseco dell’iter logico seguito dall’Autorità amministrativa[53].
Al contrario, il controllo del giudice sui giudizi tecnici si è concretizzato “nella verifica diretta dell’attendibilità delle operazioni tecniche sotto il profilo della loro correttezza, quanto a criterio tecnico e procedimento applicativo”[54], tramite l’uso delle regole e delle conoscenze tecniche appartenenti alla scienza specialistica applicata dalla p.A.
Acclarata la natura “intrinseca” del sindacato giurisdizionale sulla discrezionalità tecnica, sono emersi dubbi sulla sua intensità.
Sul punto, è sorta la dicotomia tra sindacato “debole”, ossia che non consente al giudice di sovrapporre (e, dunque, di sostituire) la propria valutazione tecnica opinabile a quella (altrettanto opinabile) della p.A.[55], e sindacato “forte”, che ammette la prevalenza del giudizio tecnico sviluppato nel processo su quello dell’Autorità amministrativa[56].
Almeno in apparenza, la giurisprudenza amministrativa ha superato questa contrapposizione con l’affermazione secondo cui il g.a., “al di là dell'ormai sclerotizzata antinomia sindacato forte/sindacato debole, deve attestarsi sulla linea di un controllo che, senza ingerirsi nelle scelte discrezionali della Pubblica autorità, assicuri la legalità sostanziale del suo agire, per la sua intrinseca coerenza anche e soprattutto in materie connotate da un elevato tecnicismo”[57].
Nell’ambito delle sanzioni antitrust (sia pure in una fattispecie[58] che intercettava il tema dei farmaci off-label), l’evoluzione del controllo giurisdizionale sulle valutazioni tecniche è giunta all’avvicendamento del sindacato di “attendibilità” con quello di “maggiore attendibilità”[59].
Ad oggi, però, permangono margini di incertezza sulla consistenza del controllo della discrezionalità tecnica rimesso al giudice amministrativo.
Premesso che questo tipo di discrezionalità è accompagnato, di frequente, da aggettivazioni – come “elevata”[60], “ampia”[61], “massima”[62] – occorre evidenziare che, non di rado, la portata del sindacato del g.a. si declina diversamente non solo in riferimento a settori-poteri differenti[63] (e, dunque, con una modulazione, per certi aspetti, fisiologica) ma – talvolta – anche nello stesso ambito-potere.
Ai fini che qui rilevano, di recente, proprio con riguardo alle valutazioni AIFA, l’autorità giurisdizionale aveva affermato che il controllo giudiziario deve attestarsi sui vizi di “manifesta” erroneità o di “evidente” illogicità del giudizio, ossia sulla “palese” inattendibilità[64].
Nella fattispecie sottoposta all’attenzione del Consiglio di Stato, quest’ultimo ha enunciato (testualmente) che il sindacato effettuato consiste in un controllo “intrinseco”, di “credibilità razionale”, di “attendibilità, coerenza e correttezza degli esiti”, per “verificare se l’autorità abbia violato il principio di ragionevolezza tecnica”.
In concreto, la pronuncia sulla nota sospensiva si colloca proprio al limite della “soglia” di sindacato indicata dallo stesso Collegio.
L’ordinanza sembra caratterizzata, infatti, da un’inversione dell’applicazione “tradizionale” del principio di precauzione[65], giustificata alla luce della (grave) situazione di emergenza e di sovraffollamento degli ospedali, per cui, nel dubbio circa la non pericolosità/piena sicurezza del farmaco per i pazienti COVID-19, il Consiglio di Stato ha ritenuto non corretta l’adozione di misure restrittive[66]. “Nell’incertezza perdurante circa l’efficacia della terapia sulla base degli standard scientifici più accreditati”[67], il g.a. ha attenzionato la potenziale efficacia del farmaco nello stadio iniziale della malattia (forse indugiando meno sul tema della pericolosità) e ha dimostrato comunque una maggiore propensione al rischio rispetto a quella dell’Agenzia.
Né sembra peregrino domandarsi, inoltre, se nella vicenda “tortuosa” sull’uso off-label dell’idrossiclorochina per la lotta al COVID-19 si insinuino anche momenti di discrezionalità amministrativa, dal momento che lo stesso Consiglio di Stato ha evocato il “bilanciamento tra gli opposti valori (quello dei medici curanti e quello tutelato da AIFA)”[68].
Da ultimo (non per importanza) occorre considerare se la nota con cui l’AIFA ha sospeso l’autorizzazione all’utilizzo di idrossiclorochina per il trattamento dell’infezione da SARS-CoV-2, al di fuori degli studi clinici, comporti – almeno indirettamente – anche un divieto totale di impiego di tale farmaco per la lotta al COVID-19.
In sede difensiva, l’AIFA ha sostenuto di aver “solo proibito il rimborso del farmaco per tale uso a carico del Servizio Sanitario Nazionale, ai sensi dell’art. 1, comma 4, del d.l. n. 356 del 1996, conv. in l. n. 648 del 1996”, ma non di aver vietato ai medici “di prescriverlo off label, sotto propria responsabilità”.
Il tenore dell’atto è effettivamente ambiguo (specie se – procedendo come il Consiglio di Stato – viene letto alla luce della successiva nota del 25 novembre[69]).
Ad ogni modo, se la nota AIFA sottende, dunque, tanto un comando (la sospensione d’autorizzazione all’uso già adottata), quanto un divieto generale (di prescrizione off-label), sorge il dubbio se quest’ultimo contenuto sia scindibile dal resto dell’atto e sia da ritenere illegittimo, più che per un difetto di istruttoria, irragionevolezza/illogicità[70], perché l’Agenzia non dispone di tale potere (visto anche quanto previsto dal già citato art. 3, comma 2, della c.d. legge Di Bella[71]).
7. Conclusioni
L’ordinanza, con cui il Consiglio di Stato ha avuto il compito – piuttosto difficile e delicato – di pronunciarsi sulla sospensione della nota AIFA, è stata accolta con “sentimenti contrastanti” [72].
Ai fini che qui rilevano, l’ordinanza è stata occasione di riflessione sul tema – sempre attuale – della discrezionalità tecnica, sull’applicazione (tutt’altro che agevole) del principio di precauzione anche nella materia dei farmaci, sul momento di bilanciamento tra interessi contrapposti, che sembra insinuarsi nella vicenda in oggetto, di cui dà conto la stessa pronuncia e sulla sussistenza (o meno) di un potere di divieto generalizzato dell’uso di un farmaco off-label in capo all’Agenzia italiana del farmaco.
Con riguardo al trattamento dell’infezione da SARS-CoV-2, peraltro, le sorti dell’idrossiclorochina restano ancora aperte.
Il Collegio ha accolto l’appello cautelare e ha sollecitato la fissazione dell’udienza di merito, ai sensi dell’art. 55, comma 10, c.p.a., “con la conseguente possibilità, in pendenza del presente giudizio, per i medici ricorrenti, come per tutti i medici abilitati ad operare sul territorio nazionale, di prescrivere l’idrossiclorochina ai pazienti affetti da SARS-CoV-2 nei primi giorni dall’esordio dei sintomi, in dosi non elevate, e in assenza di particolari controindicazioni o effetti collaterali per il singolo paziente, salve ulteriori prescrizioni di AIFA sulla scorta di ulteriori studi e aggiornamenti sui dati a sua disposizione, all’esito di più compiuta istruttoria, nella scheda dedicata all’idrossiclorochina sul sito www.aifa.gov.it, ad oggi aggiornata al 25 novembre 2020”.
Nell’ordinanza, il Collegio ha anche evidenziato che l’“AIFA provvederà ad aggiornare la scheda dell’idrossiclorochina in modo tale che essa non si presti ad essere nemmeno interpretata, per il futuro, nel senso di un assoluto divieto al suo utilizzo nei confronti dei medici”.
Il 22 dicembre 2020, l’Agenzia ha aggiornato la scheda sull’uso di questo farmaco nei pazienti adulti e ha evidenziato che “Alla luce delle evidenze che si sono progressivamente accumulate nell’uso terapeutico e su pazienti ricoverati e che dimostrano la completa mancanza di efficacia a fronte di un aumento di eventi avversi, seppur non gravi, AIFA non raccomanda l’uso dell’idrossiclorochina nei pazienti con COVID-19 in ospedale. (…) Nei pazienti con infezione da SARS-CoV-2 gestiti a domicilio, di bassa gravità e nelle fasi iniziali della malattia, esistono evidenze più limitate che dimostrano la completa mancanza di efficacia a fronte di un aumento di eventi avversi, seppur non gravi, pertanto AIFA non raccomanda l’utilizzo dell’idrossiclorchina. Una eventuale prescrizione nei singoli casi si configurerebbe quindi come uso off label”[73].
L’aggiornamento dell’AIFA si sostanzia, dunque, in una raccomandazione che – proprio per la sua natura non vincolante – non è assimilabile a un “assoluto divieto”, ma potrebbe essere motivo di riflessione (che, per materia, esula dalla presente sede) sulle possibili conseguenze in punto di responsabilità (civile e penale) del medico curante che prescriva l’idrossiclorochina per i pazienti infettati da SARS-CoV-2.
***
[1] Proprio per il suo carattere globale, l’infezione da COVID-19 (rectius, da SARS-CoV-2) è stata definita ufficialmente quale pandemia dal Direttore generale dell’Organizzazione mondiale della sanità (OMS), Tedros Adhanom Ghebreyesus, in occasione del briefing di Ginevra dell’11 marzo 2020. Tale qualificazione è l’epilogo dell’escalation nella valutazione del rischio compiuta dall’OMS che, in un primo momento, ossia lo scorso 30 gennaio 2020, ha dichiarato l’epidemia in questione “emergenza internazionale di salute pubblica”, per poi evidenziarne, il 28 febbraio 2020, il livello di minaccia “molto alto”.
[2] Si v. http://www.salute.gov.it/portale/nuovocoronavirus/dettaglioFaqNuovoCoronavirus.jsp?lingua=italiano&id=249.
[3] Con la precisazione che, secondo l’art. 2, d.lgs. 24 giugno 2003, n. 211 (“Attuazione della Direttiva 2001/20/CE relativa all’applicazione della buona pratica clinica nell’esecuzione delle sperimentazioni cliniche di medicinali per uso clinico”), per sperimentazione clinica si intende “qualsiasi studio sull’uomo finalizzato a scoprire o verificare gli effetti clinici, farmacologici e/o altri effetti farmacodinamici di uno o più medicinali sperimentali, e/o a individuare qualsiasi reazione avversa ad uno a più medicinali sperimentali, e/o a studiarne l’assorbimento, la distribuzione, il metabolismo e l’eliminazione, con l’obiettivo di accertarne la sicurezza e/o l’efficacia, nonché altri elementi di carattere scientifico e non. Questa definizione include le sperimentazioni cliniche effettuate in un unico centro o in più centri, solo in Italia o anche in altri Stati membri dell’Unione europea”.
[4] “L’idrossiclorochina (Plaquenil® cp da 200 mg o corrispondente generico) è un analogo della clorochina chimicamente molto simile e che ne condivide il meccanismo d’azione. È un antimalarico, attualmente utilizzato nel nostro Paese in campo reumatologico alla dose di 200 mg x 2 anche per periodi molto prolungati; esiste quindi ampia esperienza clinica (superiore rispetto alla clorochina) riguardo alla sua tollerabilità”. Così l’AIFA, Idrossiclorochina nella terapia dei pazienti adulti con COVID-19, 22 luglio 2020, reperibile sul sito dell’Autorità (www.aifa.gov.it).
[5] L’istituzione dell’AIFA è avvenuta con l’art. 48, co. 2, decreto-legge 30 settembre 2003, n. 269 (“Disposizioni urgenti per favorire lo sviluppo e per la correzione dell'andamento dei conti pubblici”), come convertito nella legge 2003, n. 326. Le competenze dell’Agenzia sono individuate, nel dettaglio, fra l’altro, nel comma 5 dell’art. 48 della citata legge istitutiva. Sui compiti dell’AIFA, si v. L. Casini, L’Agenzia italiana del farmaco: ufficio-agenzia o agenzia-ente pubblico?, in Giorn. dir. amm., 2004, 2, 121 ss.; M. Clarich, B.G. Mattarella, L’Agenzia del farmaco, in G. Fiorentini (a cura di), I servizi sanitari in Italia, Bologna, 2004, 263 ss.; V. Molaschi, Osservazioni sul ruolo dell’Agenzia italiana del farmaco (AIFA) nel governo della spesa farmaceutica, in Foro amm.-TAR, 2006, 233 ss.; G. Bobbio L’Agenzia italiana del farmaco, in G. Bobbio, M. Morino (a cura di), Lineamenti del diritto sanitario, Padova, 2010, 69 ss.; M. Atripaldi, L'Agenzia italiana del farmaco (AIFA) tra tutela del diritto alla salute ed esigenze finanziarie nel settore farmaceutico, in Federalismi.it, 26 luglio 2017; M. Monteduro, Modelli organizzativi e funzione: Il caso dell'Agenzia italiana del farmaco, Torino, 2018.
[6] Oltre alla nota precedente, si v. www.agenziafarmaco.gov.it. Per un approfondimento, si rinvia a P. Mighetti, M. Marchetti, Legislazione farmaceutica, Milano, 2015, 139 ss.
[7] In tema, Cons. Stato, Sez. III, 15 luglio 2019, n. 4967.
[8] La CGUE, 29 marzo 2012, in C-185/10, Commissione vs Polonia, ha affermato che, ai sensi dell’art. 6 della direttiva 2001/83/CE ss.mm.ii., la commercializzazione dei medicinali sul mercato dell’Unione europea è subordinata al conseguimento dell’AIC, rilasciata dalle autorità nazionali competenti, oppure a un'autorizzazione a norma del regolamento 2309/9/CEE. Per un approfondimento, si v. anche B. Bertanini, Tutela della salute, principio di precauzione e mercato del medicinale, Torino, 2016 (in particolare, 134 ss.).
[9] Sul tema, si rinvia anche ad A. Pira, L’uso off-label dei medicinali: un’agenda per il dopo-Covid-19, in quotidianosanità.it, 9 aprile 2020; G. Guerra, La commercializzazione dei farmaci a confronto con gli usi off-label: il difficile bilanciamento tra tutela della salute e concorrenza, in Politiche sanitarie, 2014, vol. 15, 2, 99 ss.; L. Pani, Off label: disciplina italiana piena di zone d'ombra, in IlSole24Ore, 21 marzo 2014; F. Massimino, Recenti interventi normativi e giurisprudenziali in materia di prescrizione dei farmaci off label, in Danno e resp., 2010, 12, 1104 ss.
[10] La sentenza della Corte cost., 12 gennaio 2011, n. 8, con nota di M. Gigante, Esigenze unitarie nella politica farmaceutica: l’uso off label dei farmaci tra principi fondamentali e riserva all’AIFA, in Giur. It., 2011, 12, 2492 ss., ha specificato che l’uso delle prescrizioni off-label non può essere regolato dal legislatore regionale. In argomento, di recente, si v. Cons. Stato, Sez. III, 15 dicembre 2020, n. 8033. Si v. anche Corte cost., 29 maggio 2014, n. 151, secondo cui un farmaco non è una valida alternativa terapeutica quando realizza “condizioni economicamente non accettabili e discriminatorie, tali da limitare l’accesso alle cure e, dunque, ledere la tutela del diritto alla salute costituzionalmente garantita”, poiché comporta un costo eccessivo per il SSN o la mancata rimborsabilità a scapito del paziente.
[11] A seguito della citata pronuncia della Corte cost., 29 maggio 2014, n. 151, l’art. 3, legge 16 maggio 2014, n. 79, in materia di stupefacenti e impiego off-label di medicinali, ha aggiunto il co. 4-bis, ai sensi del quale, in presenza di una alternativa terapeutica valida, previa valutazione AIFA, nell’elenco possono essere inseriti medicinali da impiegare per una indicazione terapeutica diversa da quella autorizzata, “purché tale indicazione sia nota e conforme a ricerche condotte nell'ambito della comunità medico-scientifica nazionale e internazionale, secondo parametri di economicità e appropriatezza”.
[12] Prima del 2003 (anno di istituzione dell’AIFA, su cui si rinvia supra, nota 5) la competenza spettava alla Commissione unica del farmaco. Con provvedimento del 17 gennaio 1997, tale Commissione ha indicato i criteri e i requisiti per l’inserimento dei farmaci nell’elenco, individuando nella stessa Commissione, nelle associazioni dei pazienti, nelle società scientifiche e negli organismi sanitari pubblici e/o privati i soggetti legittimati ad attuare la proposta e a presentare la documentazione necessaria per l’ammissione. Con provvedimento del 20 luglio 2000, la Commissione appena indicata ha istituito l’elenco delle specialità medicinali erogabili a totale carico del SSN, integrato e/o modificato periodicamente dall’AIFA.
[13] Sul noto caso c.d. Di Bella, si v. Corte cost., 26 maggio 1998, n. 185; sul più recente caso c.d. Stamina, si v. Id., 5 dicembre 2014, n. 274. In argomento, si v. anche Corte cost., 13 giugno 2000, n. 188.
[14] “La normativa del 1998, quindi, limita, ma non vieta la prescrizione dei medicinali fuori indicazione, con un’impostazione che si concilia con quella che è stata successivamente adottata a livello comunitario dalla Direttiva 2001/83/CE”. Così F. Massimino, Recenti interventi normativi e giurisprudenziali in materia di prescrizione dei farmaci off label, cit., 1106.
[15] Secondo la Cass. civ., 17 aprile 2019, n. 10719, “il diritto alla fruizione di prestazioni sanitarie a carico del Servizio sanitario, garantito dalla Costituzione e dall’art. 35 della Carta dei diritti fondamentali dell’Unione Europea, in favore di un numero quanto più ampio possibile di fruitori, deve dunque essere accertato sulla base dei seguenti criteri: a) le prestazioni richieste devono presentare, per specifiche condizioni cliniche o di rischio, evidenze scientifiche di un significativo beneficio in termini di salute, a livello individuale o collettivo, validate da parte della comunità scientifica; b) l’appropriatezza, che impone che vi sia corrispondenza tra la patologia e il trattamento secondo un criterio di stretta necessità, tale da conseguire il migliore risultato terapeutico con la minore incidenza sulla qualità della vita del paziente; c) l’economicità nell’impiego delle risorse, che richiede di valutare la presenza di altre forme di assistenza, meno costose ma di efficacia comparabile, volte a soddisfare le medesime esigenze ed erogabili dalle strutture pubbliche o convenzionate”.
[16] A proposito delle fasi della sperimentazione, si v. AIFA, Come nasce un farmaco, in www.aifa.gov.it: “Ha inizio con lo studio di fase 1 la sperimentazione del principio attivo sull’uomo che ha lo scopo di fornire una prima valutazione della sicurezza e tollerabilità del medicinale. Se il farmaco dimostra di avere un livello di tossicità accettabile rispetto al beneficio previsto (profilo beneficio/rischio) allora può passare alle successive fasi della sperimentazione. Nello studio di fase 2 (definito anche terapeutico-esplorativo) comincia ad essere indagata l’attività terapeutica del potenziale farmaco, cioè la sua capacità di produrre sull’organismo umano gli effetti curativi desiderati. Questa fase serve inoltre a comprendere quale sarà la dose migliore da sperimentare nelle fasi successive, e determinare l’effetto del farmaco in relazione ad alcuni parametri (come, ad esempio, la pressione sanguigna) considerati indicatori della salute del paziente. Questa seconda fase è utile quindi a dimostrare la non tossicità e l’attività del nuovo principio attivo sperimentale. Ci sono però ancora altri quesiti a cui bisogna dare una risposta: ma il farmaco quanto è efficace? Ha qualche beneficio in più rispetto a farmaci simili già in commercio? E qual è il rapporto tra rischio e beneficio? A tutte queste domande si risponde con lo studio di fase 3 (o terapeutico-confermatorio). In questo caso non sono più poche decine i pazienti “arruolati”, ma centinaia o migliaia. L’efficacia del farmaco sui sintomi, sulla qualità della vita o sulla sopravvivenza è confrontata con un placebo (sostanza priva di efficacia terapeutica), con altri farmaci già in uso, o con nessun trattamento”.
[17] Ibidem.
[18] Ibidem. L’A. prosegue evidenziando che “Deve quindi ritenersi che la norma del 2007 rappresenti una restrizione rispetto alla disciplina previgente, che deve quindi considerarsi tacitamente abrogata, limitatamente all’aspetto delle pubblicazioni scientifiche come requisito sufficiente per l’ammissibilità della prescrizione off label”.
[19] “Con decreto del Ministro della salute, da adottarsi entro centoventi giorni dalla data di entrata in vigore del presente decreto, tenuto conto anche delle linee guida EMEA per l'uso compassionevole dei medicinali, sono stabiliti i criteri e le modalità per l'uso di medicinali privi di AIC in Italia, incluso l'utilizzo al di fuori del riassunto delle caratteristiche del prodotto autorizzato nel paese di provenienza e l'uso compassionevole di medicinali non ancora registrati. Fino alla data di entrata in vigore del predetto decreto ministeriale, resta in vigore il decreto ministeriale 8 maggio 2003, pubblicato sulla Gazzetta Ufficiale della Repubblica italiana n. 173 del 28 luglio 2003”.
[20] La norma è poi confluita – con alcune modifiche/aggiunte – nell’art. 40, del decreto-legge 8 aprile 2020, n. 23 (recante “Misure urgenti in materia di accesso al credito e di adempimenti fiscali per le imprese, di poteri speciali nei settori strategici, nonché interventi in materia di salute e lavoro, di proroga di termini amministrativi e processuali”) come convertito dalla legge 5 giugno 2020, n. 40
[21] Per come, a sua volta modificato, dalla legge 11 gennaio 2018, n. 3.
[22] Come evidenziato da E. Bellomo, L’emergenza sanitaria Covid-19: l’impatto della decretazione d’urgenza sulla sperimentazione di farmaci ad uso compassionevole, in Ceridap, 26 ottobre 2020, “In questo caso, non si attua infatti quel concorso di azioni che partono dal promotore dello studio e che arrivano a coinvolgere lo sperimentatore e la struttura sanitaria lasciando che sia, invece, il medico a ritenere di dover applicare un protocollo di studio previa (secondo la normativa emergenziale) avvallo scientifico e monitoraggio di AIFA e del Comitato etico nazionale”.
[23] AIFA, COVID-19: precisazioni su definizioni di uso compassionevole e relative applicazioni del decreto legge 18/2020, 26 marzo 2020, ha specificato che la disposizione di cui all’art. 17 si applica unicamente alle richieste che ricadono nei programmi di uso terapeutico. Con tale definizione si intende “il protocollo clinico predefinito e identico per tutti i pazienti, presentato dalle aziende farmaceutiche, con applicazione di criteri univoci di inclusione, esclusione e schema di trattamento per specifici farmaci somministrati a più pazienti (secondo il DM 7/9/2017)”. Di contro, “Gli usi terapeutici nominali (…) NON devono essere sottoposti per valutazione al Comitato Etico unico Spallanzani, ma restano assoggettati alla normativa vigente e quindi rimangono di competenza dei Comitati Etici locali”. Si specifica inoltre che per la presentazione all’AIFA dei programmi di uso terapeutico su COVID-19 da parte di aziende farmaceutiche viene fatta deroga al termine dei 15 giorni antecedenti l’avvio degli stessi. L’Autorità ha chiarito, inoltre, che “Per i programmi di uso terapeutico, visti i tempi brevissimi dell'attivazione dei trattamenti d'emergenza, è consentita l'importazione di stock dei farmaci inclusi nei programmi di uso compassionevole, sulla base dei seguenti documenti: del parere preliminare favorevole dell’AIFA, oppure del parere favorevole del Comitato Etico nazionale per l'emergenza COVID Spallanzani, oppure del Comitato Etico della struttura trattante nel caso di programma già con parere favorevole da parte del relativo Comitato Etico prima del 17 marzo, oppure del Comitato Etico della struttura trattante nel caso di usi terapeutici su base nominale. Tutti i trattamenti (di cui alla definizione 1 o 2) che hanno ricevuto un parere positivo saranno pubblicati sul sito istituzionale dell’AIFA, nell’apposita sezione Emergenza COVID-19”.
[24] Si v. la Circolare sulle procedure semplificate per gli studi e gli usi compassionevoli per l'emergenza da COVID-19, 19 aprile 2020, pubblicata sul sito dell’AIFA (www.aifa.gov.it).
[25] Nel comunicato AIFA, emergenza COVID-19: costituita “Unità di crisi Coronavirus”, 12 marzo 2020, si legge che “In considerazione del fatto che nell’emergenza gli ospedali fanno ricorso a protocolli che prevedono l’uso off label di medicinali in commercio in Italia, AIFA sta predisponendo l’approvazione di quelli già identificati, che verranno sottoposti a valutazione da parte del CTS”.
[26] Si v. quanto indicato supra, par. 2.
[27] AIFA, Azioni intraprese per favorire la ricerca e l’accesso ai nuovi farmaci per il trattamento del COVID-19, 17 marzo 2020.
[28] AIFA, Comunicazione AIFA sull’utilizzo di Clorochina e Idrossiclorochina nella terapia dei pazienti affetti da COVID-19 - Informazioni di sicurezza, 31 marzo 2020.
[29] EMA, Aggiornamento sui trattamenti e i vaccini in fase di sviluppo contro il COVID-19, 31 marzo 2020.
[30] EMA, COVID-19: clorochina e idrossiclorochina devono essere utilizzati solo negli studi clinici o nei programmi di utilizzo in emergenza, 1° aprile 2020.
[31] Si v., ad es., AIFA, COVID-19 - AIFA autorizza nuovo studio clinico sull’idrossiclorochina, 9 aprile 2020.
[32] EMA, COVID-19: si richiama nuovamente l’attenzione sul rischio di gravi effetti indesiderati con clorochina e idrossiclorochina, 23 aprile 2020.
[33] AIFA, COVID-19 - Aggiornamento scheda informativa AIFA su idrossiclorochina, 29 aprile 2020.
[34] AIFA, AIFA sospende l’autorizzazione all’utilizzo di idrossiclorochina per il trattamento del COVID-19 al di fuori degli studi clinici, 26 maggio 2020.
[35] AIFA, COVID-19. Le motivazioni della decisione AIFA sull'uso di idrossiclorochina e clorochina, 29 maggio 2020.
[36] EMA, COVID-19: ribaditi i rischi di clorochina e idrossiclorochina, 29 maggio 2020.
[37] L’OMS ha poi ripreso la sperimentazione a inizio giugno per interromperla il 17 giugno 2020. Si v. WHO, Coronavirus disease (COVID-19): hydroxycloroquine, 19 giugno 2020 (pubblicato sul sito www.who.int).
[38] Con l’aggiunta che, a fine novembre 2020, il comitato per la sicurezza (Prac) dell'EMA ha raccomandato di aggiornare le informazioni sui farmaci contenenti clorochina o idrossiclorochina, proprio perchè la revisione di tutti i dati disponibili ha confermato un collegamento tra l'uso di questi medicinali e il rischio di disturbi psichiatrici e di comportamento suicidario. Si v. Redazione ANSA, Ema, rischio suicidio collegato a uso idrossiclorochina, in Ansa.it, 30 novembre 2020.
[39] MR Mehra e a., RETRACTED: Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis, in The Lancet, pubblicato il 22 maggio 2020 e ritirato il 5 giugno 2020.
[40] AIFA, Idrossiclorochina nella terapia dei pazienti adulti con COVID-19, Update del 22 luglio 2020. Si v. anche l’aggiornamento, AIFA, COVID-19: AIFA limita l’uso di remdesivir in casi selezionati e consente idrossiclorochina solo in studi clinici randomizzati a domicilio, 26 novembre 2020, con cui l’Autorità ha confermato la sospensione dell’autorizzazione all’utilizzo off-label dell’idrossiclorochina “sia per l’uso terapeutico (ospedaliero e territoriale) sia per l’uso profilattico, sulla base delle evidenze che si sono progressivamente accumulate e che dimostrano la completa mancanza di efficacia a fronte di un aumento di eventi avversi, seppur non gravi”. Ha poi soggiunto che “L’utilizzo nei pazienti non gravi e nelle fasi iniziali della malattia può essere consentito solo nell’ambito di studi clinici randomizzati, in quanto al momento le evidenze, seppur tendenzialmente negative, sono ancora limitate”.
[41] E. Burba, È iniziata la battaglia dell’idrossiclorochina, in Panorama, 24 novembre 2020; S. Turina, Covid e idrossiclorochina: che cosa dicono gli ultimi pareri scientifici. Le ragioni di una bocciatura, in Corriere della sera, 12 dicembre 2020.
[42] Si pensi allo studio promosso dal Movimento IppocrateOrg, intitolato “Recovery trial and hydroxychloroquine”, pubblicato sull’International Medical Journal, il 29 Settembre 2020. Si v. anche C. Scaldaferri, Chi usa e chi vieta l'idrossiclorochina nella lotta al Covid, in Agi, 18 maggio 2020.
[43] Avverso l’ordinanza del TAR Lazio, Roma, Sez. III-quater, 16 novembre 2020, n. 7069.
[44] Il TAR Lazio, Roma, Sez. III-quater, con l’ordinanza (non appellata) del 14 settembre 2020, n. 5911, ha rigettato l’istanza cautelare avverso la nota AIFA del 26 maggio 2020.
[45] A sostegno di tale interpretazione, il Cons. Stato ha invocato la determinazione n. 484 del 2020, pubblicata in G.U. n. 112 del 2 maggio 2020, e l’aggiornamento (citato supra, nota 39) al 25 novembre 2020 della scheda sull’idrossiclorochina del 25 novembre 2020.
[46] Si v. anche l’art. 1 c.p.a.
[47] Sul tema, tra i numerosi contributi, si v. F. Cammeo, La competenza di legittimità della IV Sezione e l’apprezzamento dei fatti valutabili secondo criteri tecnici, in Giur. it., 1902, III, 276 ss.; E. Presutti, Discrezionalità pura e discrezionalità tecnica, ivi, 1910, 16 ss.; Id., I limiti del sindacato di legittimità, Milano, 1911; M.S. Giannini, Il potere discrezionale della pubblica amministrazione. Concetto e problemi, Milano, 1939, ora in Id., Scritti, Milano, 2000, I, 387 ss.; P. Virga, Appunti sulla cosiddetta discrezionalità tecnica, in Jus, 1957, 95 ss.; V. Bachelet, L’attività tecnica della pubblica amministrazione, Milano, 1967; F. Ledda, Potere, tecnica e sindacato giudiziario sull’amministrazione pubblica, in Dir. proc. amm., 1983, 372 ss. (e in Studi in memoria di Vittorio Bachelet, Milano, 1987, II); V. Cerulli Irelli, Note in tema di discrezionalità amministrativa e sindacato di legittimità, in Dir. proc. amm., 1984, 463 ss.; A.M. Sandulli, Manuale di diritto amministrativo, Napoli, 1989, 575 ss.; C. Marzuoli, Potere amministrativo e valutazioni tecniche, Milano, 1985; F. Salvia, Attività amministrativa e discrezionalità tecnica, in Dir. proc. amm., 1992, 685 ss.; G. Pelegatti, Valutazioni tecniche dell’amministrazione pubblica e sindacato giudiziario, un’analisi critica dei recenti sviluppi della dottrina giuspubblicistica, in Riv. trim. dir. pubbl., 1992, 158 ss.; D. De Pretis, Valutazione amministrativa e discrezionalità tecnica, Padova, 1995; N. Paolantonio, Interesse pubblico ed apprezzamenti amministrativi, in Dir. amm., 1996, 2, 413 ss.; Id., Il sindacato di legittimità sul provvedimento amministrativo, Padova, 2000; B. Tonoletti, L’accertamento amministrativo, Padova, 2001; F. Fracchia, C. Videtta, La tecnica come potere, in Foro amm., 2002, III, 493 ss.; D. Mastrangelo, La tecnica nell’amministrazione fra discrezionalità pareri e merito, Bari, 2003; A. Travi, Il giudice amministrativo e le questioni tecnico-scientifiche: formule nuove e vecchie soluzioni, in Dir. pubbl., 2004, 2, 439 ss.; F. Cintioli, Giudice amministrativo, tecnica e mercato. Poteri tecnici e giurisdizionalizzazione, Milano, 2005; Id., Tecnica e processo amministrativo, in Dir. proc. amm., 2004, 4, 983 ss.; Id., Discrezionalità tecnica (voce), in Enc. del diritto, II, 2009, 471 ss.; A. Giusti, Contributo allo studio di un concetto ancora indeterminato. La discrezionalità tecnica della pubblica amministrazione, Napoli, 2007; M. Allena, Il sindacato del giudice amministrativo sulle valutazioni tecniche complesse: orientamenti tradizionali versus obblighi internazionali, in Dir. proc. amm., 4, 2012, 1602 ss.; F. Volpe, Il sindacato sulla discrezionalità tecnica tra vecchio e nuovo rito (considerazioni a margine della sentenza Cass. SS. UU., 17 febbraio 2012, n. 2312), in Giustamm.it, 2012. Si consideri, inoltre, la recente Ricerca dell’Ufficio Studi, curata dal Presidente M. Lipari, dai Cons. Giovagnoli e Storto, con la collaborazione dei Presidenti R. De Nictolis e R. Chieppa, Autorità indipendenti di regolazione dei mercati e tutela giurisdizionale amministrativa, in www.giustizia amministrativa.it, 2019.
[48] Così il TAR Trentino-Alto Adige, ordinanza 5 maggio 2008, n. 228; M.S. Giannini, Istituzioni di diritto amministrativo, Milano, 2000, 269; Id., Diritto amministrativo, vol. II, Milano, 1993, 55-56 ha affermato che la discrezionalità tecnica “non ha proprio nulla di discrezionale” e che, quindi, tale definizione costituisce “un errore storico della dottrina”; come ha evidenziato E. Capaccioli, citato in P. Lazzara, L'opera scientifica di Enzo Capaccioli tra fatto, diritto e teoria generale, in Dir. amm., 2009, 995 ss., § 4, “le due valutazioni tecnica e discrezionale, non coincidono mai; dove comincia l'una finisce l'altra e dove c'è l'una non vi è posto per l'altra”. Si v. anche F. Merusi, La Teoria generale di Enzo Capaccioli nel dibattito amministrativo contemporaneo, in Dir. amm., 2009, 873 ss. Nello stesso senso, F. Salvia, Attività amministrativa e discrezionalità tecnica, cit., 688-689, ha sottolineato l’ “equivocità della formula”.
[49] Su cui si v. M.S. Giannini, Diritto amministrativo, II, Milano, 1988, 54.
[50] A tale riguardo, R. Villata, M. Ramajoli, Il provvedimento amministrativo, Milano, 2017, 179, hanno evidenziato che la formulazione di un giudizio “alla stregua di una scienza, in luogo di legittimare una riserva di amministrazione, costituisce all’opposto la premessa della sua verificabilità”. Contra, si v. la posizione di G. Clemente Di San Luca, Il sindacato giurisdizionale sulle valutazioni tecniche in materia ambientale, in Giustamm.it, 7, 2016.
[51] A partire da Cons. Stato, Sez. IV, 9 aprile 1999, n. 601, in Foro it., 2001, III, 11, con nota di A. Travi.
[52] Oggi disciplinata dagli artt. 63, comma 4 e 67 c.p.a. Come evidenziato da A. Travi, Sindacato debole e giudice deferente, in Giorn. dir. amm., 2006, 3, 313: “Nel giudizio le parti sono sempre sullo stesso piano e l’attendibilità dell’affermazione di una parte non può essere privilegiata per il fatto che… sia stata recepita in un provvedimento amministrativo (art.113 Cost). La regola del giudizio è sempre la stessa: tutti i fatti controversi devono essere verificati nel rispetto delle norme sulle allegazioni e sulla prova e tale regola non incontra eccezioni nella dimensione di ordine tecnico dei fatti: altrimenti la giustizia amministrativa diventa qualcosa di diverso e minore rispetto alla giustizia senza aggettivi”.
[53] Si veda anche Cass., Sez. Un., 20 gennaio 2014 n. 1013, in Dir. proc. amm. 2014, 1057, con nota di B. Gilberti: “l’esercizio della discrezionalità tecnica, non essendo espressione di un potere di supremazia della pubblica amministrazione, non è di per sé solo idoneo a determinare l’affievolimento dei diritti soggettivi di coloro che del provvedimento amministrativo siano eventualmente pregiudicati. Non può pertanto sostenersi che chi lamenti la lesione del proprio diritto, a causa del cattivo esercizio della discrezionalità tecnica, non possa chiederne l’accertamento al giudice, il quale non potrà quindi esimersi dal verificare se le regole della buona tecnica sono state o meno violate dall’amministrazione. Ne fornisce evidente conferma il fatto stesso che il giudice amministrativo disponga oggi di ampi mezzi istruttori, ivi compreso lo strumento della consulenza tecnica”.
[54] Così, di recente, Cons. Stato, Sez. I, 30 novembre 2020, n. 1958.
[55] A tale fine, A. Romano Tassone, Sulle vicende del concetto di «merito», in Dir. amm., 2008, 549, ha evocato la nozione di “preferenza” – anziché di “riserva” – di amministrazione, nel senso che la valutazione ragionevole, ancorchè opinabile, dell’autorità amministrativa deve essere preferita rispetto alle altre possibili alternative “ragionevoli e legittime”.
[56] Secondo G. Clemente Di San Luca, Il sindacato giurisdizionale sulle valutazioni tecniche in materia ambientale, cit., “Anche questa distinzione è da considerarsi priva di senso: l’unico sindacato coerente con il principio di separazione delle funzioni resta quello basato sul vizio dell’eccesso di potere. E tale vizio – una volta essendo stato acquisito che può risolversi, oltre che nella forma del mero sviamento, pure in quella fondata sulla rilevazione delle figure sintomatiche – ben può rivelarsi sussistente anche laddove la valutazione compiuta dalla P.A., alla verifica della sua attendibilità tecnico-scientifica, risulti superficiale, incongrua, irragionevole, inadeguata, ecc. Il che, evidentemente, non può che integrare una delle varie figure sintomatiche progressivamente elaborate, in un tempo ormai assai lungo, dalla giurisprudenza amministrativa: non diversamente da quanto accade con riguardo alla valutazione che la P.A. compie in funzione della scelta del contenuto provvedimentale”.
[57] Cons. Stato, Sez. III, 25 marzo 2013, n. 1645. In termini, di recente, TAR Lombardia, Milano, Sez. II, 25 febbraio 2019, n. 408.
[58] Ossia il noto caso La Roche-Novartis (o Avastin-Novartis).
[59] Cons. Stato, Sez. VI, 19 luglio 2019, n. 4990, in Foro it., 2019, 10, 533 ss. con nota di R. Pardolesi; in Giustamm.it, con nota di G. Cice. Nell’Osservatorio sulla giustizia amministrativa, in Foro amm., 2019, 9, 1377 ss., è evidenziato che il Cons. Stato, sez. VI, 5 agosto 2019, da 5558 a 5564 e 6022, 6023, 6025, 6027, 6030, 6032 e 6065; Id., 23 settembre 2019, n. 6314, hanno richiamato il Cons. Stato, Sez. VI, 19 luglio 2019, n. 4990, ma limitatamente “alla parte ricognitiva delle precedenti acquisizioni (sindacato “non sostitutivo di attendibilità”), estromettendo quella che ha proposto il revirement in senso ampliativo (sindacato “pieno di maggiore attendibilità”)”. Sul tema, si v. R. Garofoli, Il controllo giudiziale amministrativo e penale della pubblica amministrazione, in Riv. trim. dir. pubbl., 2020, 2, 405 ss.; F. Goisis, L’efficacia di accertamento autonomo del provvedimento AGCM: profili sostanziali e processuali, in Dir. proc. amm,, 2020, 1, 45 ss.; S. Torricelli, Per un modello generale di sindacato sulle valutazioni tecniche: il curioso casi degli atti delle autorità amministrative indipendenti, in Foro amm., 2020, 1, 97 ss.; M. Cappai, Il problema del sindacato giurisdizionale sui provvedimenti dell’AGCM in materia antitrust: un passo in avanti, due indietro … e uno in avanti. Una proposta per superare l’impasse, in Federalisimi.it, 2019. Per F. Patroni Griffi, Giustizia amministrativa: evoluzione e prospettive nell’ordinamento nazionale e nel quadro europeo, in www.giustizia-amministrativa.it, 2020, “in settori tradizionali (per esempio, quello dei beni culturali o dei concorsi universitari, mentre resiste quello degli esami di abilitazione) o di più recente rilevanza (soprattutto quello delle sanzioni e della regolazione economica, sia pure con accenti che devono restare differenti), l’area del “merito” amministrativo resta confinata alla scelta vera e propria, mentre il giudice valuta se la scelta effettuata in concreto sia quella dotata di “maggiore attendibilità” e non semplicemente quella comunque riconducibile al novero delle opzioni possibili. Con il solo ovvio limite della sostituzione di una propria scelta a quella amministrativa. È chiaro che questo principio riuscirà ad affermarsi se i giudici amministrativi saranno particolarmente sensibili alle evenienze in fatto del processo e la Corte di cassazione saprà distinguere, in sede di controllo sui limiti cd. esterni della giurisdizione amministrativa, tra area riservata al merito amministrativo e sindacato pieno ed effettivo sulla legittimità dell’azione amministrativa.”. Sull’applicabilità del criterio della maggiore attendibilità la giurisprudenza non è univoca. Ad esempio, con riferimento al vincolo culturale apposto dalla p.A., il TAR Piemonte, Sez. I, 3 marzo 2020, n. 155, ha sostenuto che “I recenti orientamenti sui limiti che il giudice incontra nel sindacato della c.d. discrezionalità tecnica impongono al Collegio di verificare se l’indagine qualitativa del bene effettuata dall’amministrazione si presenti come quella dotata di maggiore attendibilità e non solo come una delle tante valutazioni possibili”; a proposito del giudizio tecnico sull’infermità da causa di servizio, il TAR Lazio, Roma, Sez. II, 1° dicembre 2020, nn. 12790 e 12794 ha affermato, invece, che “il sindacato del giudice amministrativo è ammissibile laddove la valutazione si ponga al di fuori dell’opinabilità o della maggiore attendibilità”.
[60] Si v., ad esempio, di recente, TAR Lazio, Roma, Sez. I, 4 settembre 2020, n. 9335.
[61] Si v., ad esempio, di recente, Cons. Stato, Sez. VI, 4 gennaio 2021, n. 37; TAR Lombardia, Milano, Sez. III, 4 gennaio 2021, n. 4.
[62] Si v., ad esempio, di recente, Cons. Stato, Sez. IV, 15 ottobre 2020, n. 6258; TAR Lazio, Roma, Sez. I-bis, 24 luglio 2020, n. 8741.
[63] F. Patroni Griffi, Giustizia amministrativa: evoluzione e prospettive nell’ordinamento nazionale e nel quadro europeo, cit.; Id., Il sindacato del Giudice amministrativo sugli atti delle Autorità Indipendenti, in www.giustizia-amministrativa.it; C. Deodato, Nuove riflessioni sull’intensità del sindacato del giudice amministrativo. Il caso delle linee guida dell’ANAC, in Federalismi.it, 2, 2017. Di recente, sul tema, su iniziativa della Prof.ssa M.A. Sandulli e del Prof. F. Francario, si è svolta una Tavola rotonda per l’apertura del modulo di diritto amministrativo della SSPL dell’Università egli studi “Roma Tre”, intitolata “Attività discrezionale e attività vincolata della PA e sindacato del GA”, Roma, 30 gennaio 2020. Si v. anche il webinar “La discrezionalità tecnica tra procedimento e processo”, organizzato dalla Città Metropolitana di Firenze, in collaborazione con l’Università degli Studi di Firenze, 25 giugno 2020.
[64] Si v. TAR Lazio, Roma, Sez. II-quater, 13 agosto 2020, n. 9198. Secondo, TAR Lazio, Roma, Sez. II-quater, 17 gennaio 2020, n. 591, 592 e 593: “quello della commissione AIFA costituisca senz’altro giudizio connotato da discrezionalità tecnica: pertanto occorre dimostrare anche l’abnormità di un siffatto giudizio onde poter demolire la eventuale determinazione finale. In altre parole, il relativo sindacato giurisdizionale deve attestarsi su riscontrati (e prima ancora dimostrati) vizi di manifesta erroneità o di evidente illogicità del giudizio stesso, ossia sulla palese inattendibilità della valutazione espressa dalla stessa commissione AIFA. A tale specifico riguardo osserva infatti il collegio che il sindacato del giudice sulla discrezionalità tecnica, quale è quello che caratterizza la valutazione di equivalenza terapeutica tra medicinali, non può sfociare nella sostituzione dell’opinione del giudice a quella espressa dall’organo dell’amministrazione ma è piuttosto finalizzato a verificare se il potere amministrativo sia stato esercitato mediante utilizzo delle regole conforme a criteri di logicità, congruità e ragionevolezza. Sicché un tale sindacato rimane limitato ai casi di macroscopiche illegittimità, quali errori di valutazione gravi ed evidenti oppure valutazioni abnormi o inficiate da errori di fatto, pena un’inammissibile invasione della sfera propria della P.A. Il parere della commissione AIFA costituisce pertanto atto di esercizio di ampia discrezionalità tecnica e il sindacato del giudice amministrativo è di tipo intrinseco debole (o di attendibilità), limitato cioè alla verifica della sussistenza di vizi sintomatici dell’eccesso di potere quali la palese carenza di istruttoria e l’abnorme travisamento dei fatti nonché la evidente illogicità e incongruenza delle valutazioni espresse”.
[65] Tale principio non è espressamente enunciato nel testo della pronuncia, ma sembra implicitamente presente, visto che, più volte, il g.a. ha evocato il concetto di “rischio” e il “rapporto rischi/beneifici”. “Si riferisce a un approccio alla gestione del rischio in base al quale, se vi è la possibilità che una data politica o azione possa danneggiare il pubblico o l’ambiente, e se non c’è ancora consenso scientifico sulla questione, la politica o l’azione in questione non dovrebbe essere perseguita”. Sul principio di precauzione, tra i numerosi contributi, si rinvia a U. Beck, Risikogesellschaft. Auf dem Weg in eine andere Moderne, Frankfurt am Main, 1986; A. Jordan, J. Cameron, Interpreting the Precautionary principle, London, 1994; U. Di Fabio, Gefahr, Vorsorge, Risiko, in Jura, 1996, 566 ss.; A. KISS, The rights and interest of future
generations and the precautionary principle, in D. Freeston, E. Hey, The Precautionary principle and international law. The challenge of implementation, The Hague, 1996, 19 ss.; J. Scott, The Precautionary Principle Before the European Courts, in R. Macrory (a cura di), Principles of European Environmental Law, Groningen, Europa, 2004; V. Heyvaert, Facing The Consequences of the Precautionary Principle in European Community Law, in European Law Review, 2006, 185 ss.; A. Barone, Il diritto del rischio, Milano, 2006; A. Bianchi, M. Gestri (a cura di), Il principio precauzionale nel diritto internazionale e comunitario, Milano, 2006; D. Bevilacqua, I limiti della scienza e le virtù della discrezionalità: il principio di precauzione nel diritto globale, in G. Della Cananea (a cura di), I principi dell’azione amministrativa nello spazio giuridico globale, Napoli, 2007; M. Antonioli, Precauzionalità, gestione del rischio ed azione amministrativa, in Riv. It. Dir. pubbl., 2007, 51 ss.; A. Zei, Il principio di precauzione: programma, regola, metodo, in R. Bifluco, A. D’Aloia, Un diritto per il futuro, Napoli, 2008; Id., Principio di precauzione, in Dig. disc. pubbl., Torino, II, 2008, 670 ss.; C.R. Sunstein, Il diritto della paura, Bologna, 2010; P. Savona, Il principio di precauzione e il suo ruolo nel sindacato giurisdizionale sulle questioni scientifiche controverse, in Federalismi.it, 2011; S. Cognetti, Potere amministrativo e principio di precauzione fra discrezionalità tecnica e discrezionalità pura, in S. Cognetti, A. Contieri, S. Licciardello, F. Manganaro, S. Perongini, F. Saitta (a cura di), Percorsi di diritto amministrativo, Torino, 2014, 142 ss.; F. De Leonardis, Il principio di precauzione, in M. Renna, F. Saitta (a cura di), Studi sui principi del diritto amministrativo, Milano, 2012, 413 ss.; Id., Tra precauzione, prevenzione e programmazione, in L. Giani, M. D’Orsogna, A Police, Dal diritto dell’emergenza al diritto del rischio, Napoli, 2018, 49 ss.; con specifico riferimento all’applicazione del principio di precauzione nell’emergenza COVID-19, si v., in particolare, F. Scalia, Principio di precauzione e ragionevole bilanciamento dei diritti nello stato di emergenza, in Federalismi.it, 32, 2020; G. Pitruzzella, Il principio di precauzione è debole nella società globale del rischio, in Il Foglio, 12 luglio 2020; M. Basigli, L’epidemia di CoVid-19: il principio di precauzione e i fallimenti istituzionali, in Mercato concorrenza regole, 2019, 3, 475 ss. Sulla natura “transettoriale” del principio di precauzione, che ormai non attiene più solo al diritto ambientale (art. 191, par. 2, TFUE), si v., tra gli altri, P. Kourilssky, G. Viney, Le principe de précaution, Parigi, 2000, 144; B. Delfino, Una nuova nozione di sicurezza nel diritto pubblico: riflessioni, rapporti con i principi di prevenzione e di precauzione e sua forza espansiva, in Foro amm., 2008, 11, 3183 ss.; F. De Leonardis, Il principio di precauzione, cit., 416-417 e la CGUE, Grande Sezione, 1° ottobre 2019, Mathiew Blaise e a., C-616/17, par. 41.
[66] Con riferimento all’applicazione del principio in materia di farmaci, si v. TAR Lazio, Roma, Sez. III-quater, 9 febbraio 2017, n. 2225, che ha affermato che l'insegnamento in materia della Corte di Giustizia dell’Unione europea, «(…) qualora risulti impossibile determinare con certezza l'esistenza o la portata del rischio asserito a causa della natura insufficiente, non concludente o imprecisa dei risultati degli studi condotti, ma persista la probabilità di un danno reale per la salute nell'ipotesi in cui il rischio si realizzasse, il principio di precauzione giustifica l'adozione di misure restrittive»”. Con riguardo alle misure adottate in fase di emergenza, si v., ad esempio, TAR Campania, Napoli, Sez. V, ordinanza 22 aprile 2020, n. 826, in www.quotidianogiuridico.it, che – nel rigettare l’stanza di sospensiva di un provvedimento con cui era stata disposta la chiusura temporanea di una casa di cura – ha richiamato il Cons. Stato, Sez. III, 3 ottobre 2019, n. 6655, secondo cui “Il c.d. « principio di precauzione », di derivazione comunitaria (art. 7, Regolamento n. 178 del 2002), impone che quando sussistono incertezze o un ragionevole dubbio riguardo all'esistenza o alla portata di rischi per la salute delle persone, possono essere adottate misure di protezione senza dover attendere che siano pienamente dimostrate l'effettiva esistenza e la gravità di tali rischi; l'attuazione del principio di precauzione comporta dunque che, ogni qual volta non siano conosciuti con certezza i rischi indotti da un'attività potenzialmente pericolosa, l'azione dei pubblici poteri debba tradursi in una prevenzione anticipata rispetto al consolidamento delle conoscenze scientifiche”)”.
[67] Così l’ordinanza in commento.
[68] Per completezza, si segnala che in un passaggio precedente dell’ordinanza, si afferma, però, che “Si deve anzitutto fugare ogni dubbio circa l’insinuarsi di un pericoloso relativismo terapeutico o irrazionalismo decisorio, fondato su nebulose intuizioni curative, più o meno verificabili, del singolo medico, su pseudoconoscenze del paziente o addirittura su valutazioni di mera opportunità politica dello stesso decisore pubblico, in quanto le decisioni sul merito delle scelte terapeutiche, in relazione alla loro appropriatezza, dovrebbero prevedere «l’elaborazione di indirizzi fondati sulla verifica dello stato delle conoscenze scientifiche e delle evidenze sperimentali acquisite, tramite istituzioni e organismi – di norma nazionali e sovra-nazionali – a ciò deputati, dato l’essenziale rilievo che a questi fini rivestono gli organi tecnico-scientifici»”.
[69] Su cui si rinvia supra.
[70] Come ritenuto dal Consiglio di Stato (con riferimento alla nota integralmente intesa).
[71] Per cui se manca (o è sospesa) l’autorizzazione AIFA, ma persistono i requisiti per la prescrizione “fuori dal bugiardino”, ai sensi di tale disposizione, il medico può comunque impiegare tale farmaco (viceversa, se non sussistono i presupposti ivi stabiliti, il sanitario non può utilizzare il farmaco, perché è la legge stessa a vietarlo).
[72] Per un commento favorevole alla pronuncia, si v., tra gli altri, l’Assessore alla sanità del Piemonte, come riportata da E. Burba, Il Piemonte dà il via libera all’idrossiclorochina, in Panorama, 16 dicembre 2020; P. Varese nell’intervista rilasciata ad A. Mariotti, L’idrossiclorochina torna tra i farmaci che si possono usare contro il Covid: “Sentenza storica per i medici”, in La stampa, 12 dicembre 2020; Contra, si v. la posizione del Presidente dei medici internisti della Fedoi, D. Manfellotto, e del Presidente del Consiglio superiore di sanità, F. Locatelli, riportate da Huffpost, Ok del Consiglio di Stato all’idrossiclorochina? I giudici vogliono sostituirsi alla scienza, in Huffingtonpost, 12 dicembre 2020; L. Simonetti, Il covid in tribunale, in Il Foglio, 18 dicembre 2020; l’editoriale G. Corbellini, Il Tribunale del Covid, ivi, 12 dicembre e G. Ciliberto, Gli studi dimostrano che l’idrossiclorochina non ha alcun beneficio contro il Covid, ivi.
[73] AIFA, Idrossiclorochina nella terapia dei pazienti adulti con COVID-19, Update del 22 dicembre 2020.

